Hypertrophic cardiomyopathy (HCM), Previously known as idiopathic hypertrophic subaortic stenosis (IHSS) or asymmetrical septal hypertrophy (ASH), is the most prevalent genetic cardiovascular disorder, arises from mutations in genes responsible for encoding cardiac sarcomere proteins, leading to diverse phenotypic expressions and clinical courses. This condition stands as the primary cause of sudden death among young individuals.
Pathophysiology
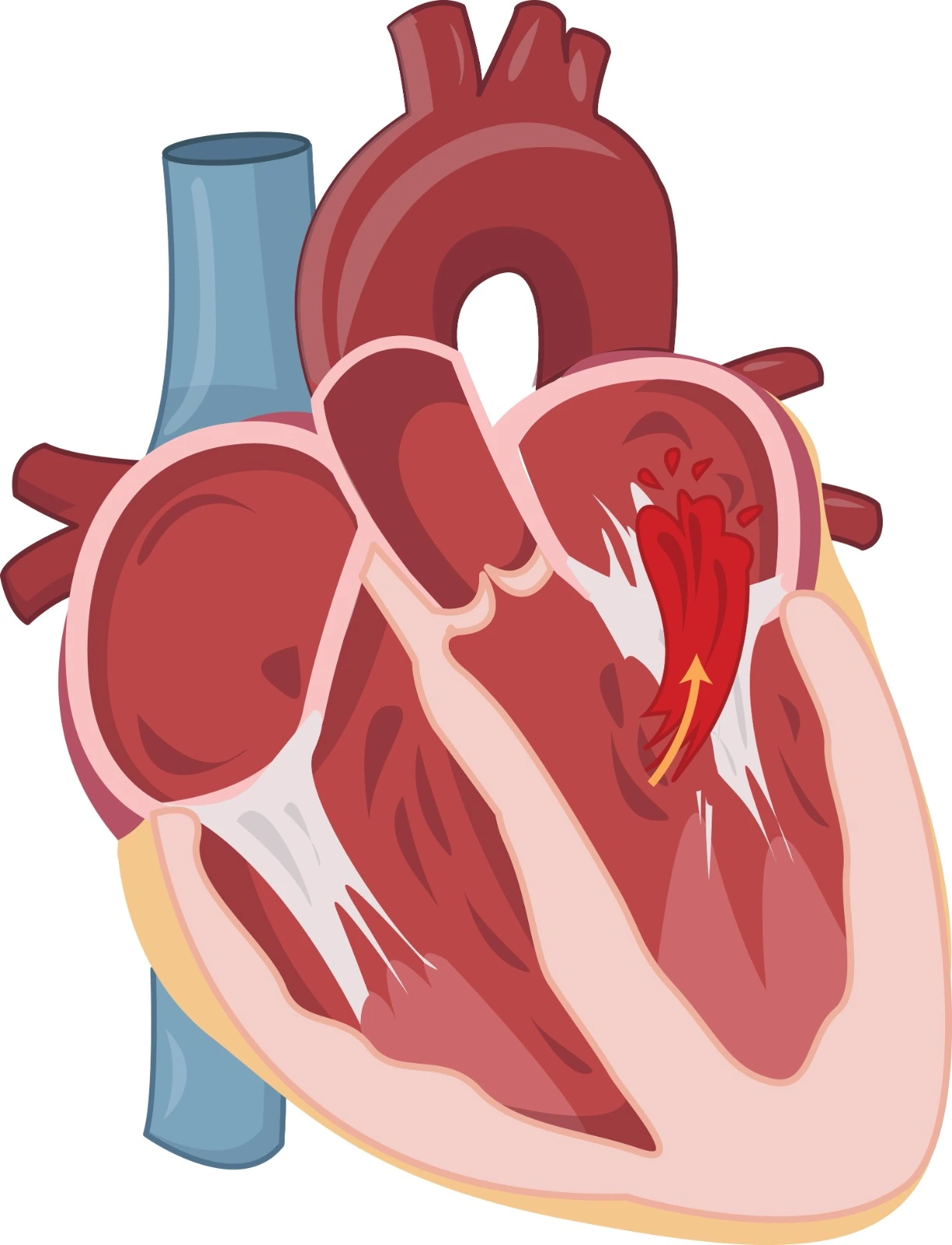
Hypertrophic cardiomyopathy (HCM) has long been characterized by a significant focus on the dynamic pressure gradient across the left ventricular (LV) outflow tract. This attention stems from its association with the further constriction of an already narrowed outflow tract, a condition exacerbated by marked asymmetrical septal hypertrophy and potentially an abnormal mitral valve position. The systolic anterior motion of the mitral valve, a key aspect in this scenario, has prompted three proposed explanations.
Firstly, it has been suggested that the contraction of papillary muscles, influenced by the abnormal location of the mitral valve and septal hypertrophy, pulls the valve against the septum. Alternatively, the abnormal position of the mitral valve in the outflow tract may push it against the septum. A third perspective attributes the motion to the Venturi effect, where the mitral valve is drawn toward the septum due to lower pressure resulting from high-velocity blood ejection through a narrowed outflow tract.
In addition to these mechanical intricacies, most HCM patients exhibit abnormal diastolic function, regardless of the presence of a pressure gradient. This impairment in ventricular filling increases filling pressure, even in cases with a normal or small ventricular cavity. Underlying these diastolic abnormalities are disturbed calcium kinetics and subendocardial ischemia, both intricately linked to the profound hypertrophy and myopathic processes characteristic of HCM.
Etiology
The etiology of hypertrophic cardiomyopathy (HCM) encompasses various factors contributing to the inappropriate myocardial hypertrophy and distinctive features associated with the condition.
Abnormal Calcium Kinetics
- Data establishes a significant link between abnormal myocardial calcium kinetics and the development of HCM, particularly in patients with diastolic functional abnormalities.
- Dysregulation of calcium fluxes, stemming from an increased number of calcium channels, results in elevated intracellular calcium concentration, potentially leading to hypertrophy and cellular disarray.
Genetic Causes
- Familial HCM, accounting for approximately 50% of cases, follows an autosomal dominant Mendelian inheritance pattern.
- Sporadic forms may arise from spontaneous mutations.
- More than 50 different mutations across at least 6 different genes on 4 chromosomes have been identified.
- Familial HCM is genetically heterogeneous, involving defects at multiple loci.
- The genetic basis was first reported by Seidman and collaborators in 1989, identifying a disease gene on the long arm of chromosome 14, later found to encode for beta cardiac myosin heavy chain.
- Phenotypic expression of gene mutations varies widely, with specific mutations associated with particular symptoms, the degree of hypertrophy, and prognosis.
Other Possible Causes
- Abnormal sympathetic stimulation, heightened heart responsiveness to catecholamines, or reduced neuronal norepinephrine uptake is a potential cause of HCM.
- Abnormally thickened intramural coronary arteries, failing to dilate normally, lead to myocardial ischemia, progressing to fibrosis and abnormal compensatory hypertrophy.
- Subendocardial ischemia, associated with cardiac microcirculation abnormalities, depletes energy stores essential for calcium sequestration during diastole, resulting in persistent interaction of contractile elements during diastole and increased diastolic stiffness.
- Cardiac structural abnormalities, such as a catenoid septum configuration, contribute to myocardial cell hypertrophy and disarray.
Clinical Presentation
Hypertrophic Cardiomyopathy (HCM) is a condition with diverse clinical manifestations. Many individuals show no symptoms or only minor ones, and diagnosis often occurs through family screening or routine examinations. Common symptoms include dyspnea, fatigue, chest pain, and syncope, but their severity may not consistently correlate with the extent of heart muscle thickening. Women may present at an older age with higher rates of symptom progression.
Heart failure symptoms, mainly dyspnea on exertion, can occur due to various mechanisms such as diastolic dysfunction and LVOT obstruction. Chest pain resembling angina is common and linked to microvascular angina. Arrhythmias, including palpitations and syncope, are frequent, and acute hemodynamic collapse, though rare, may happen in patients with LVOT obstruction.
Approximately 5 percent may progress to end-stage HCM with adverse LV remodeling. Physical examination reveals systolic murmurs, and other findings like brisk pulses and a forceful LV apical impulse. Despite diverse presentations, there isn't always a predictable correlation between symptoms and obstruction degree. Regular follow-up and individualized management are essential for addressing HCM's varied manifestations.
Diagnosis
Start with a comprehensive history and physical examination.
Use multiple tests for diagnosis, severity assessment, and prognosis.
Laboratory Studies
- Include hemoglobin, fasting glucose, renal and liver function tests, BNP, NT-proBNP, troponin T, and thyroid function tests.
- Additional tests based on specialist evaluation.
Two-Dimensional Echocardiography (TTE):
- Essential for HCM diagnosis.
- Evaluates cardiac morphology, LV function, outflow tract gradient, and mitral regurgitation.
LV Hypertrophy (LVH) Assessment
- Evaluated using parasternal short-axis images.
- Unexplained LV wall thickness ≥15 mm confirms HCM.
- SAM of the mitral valve in LVOT can lead to obstruction.
Ambulatory ECG Monitoring
- Important for assessing ventricular arrhythmias and sudden cardiac death risk.
- Performed for 24-48 hours.
Cardiac Magnetic Resonance Imaging (CMRI)
- Considered when HCM diagnosis is uncertain after echocardiography.
- Provides detailed information on hypertrophy, septal anatomy, and fibrosis.
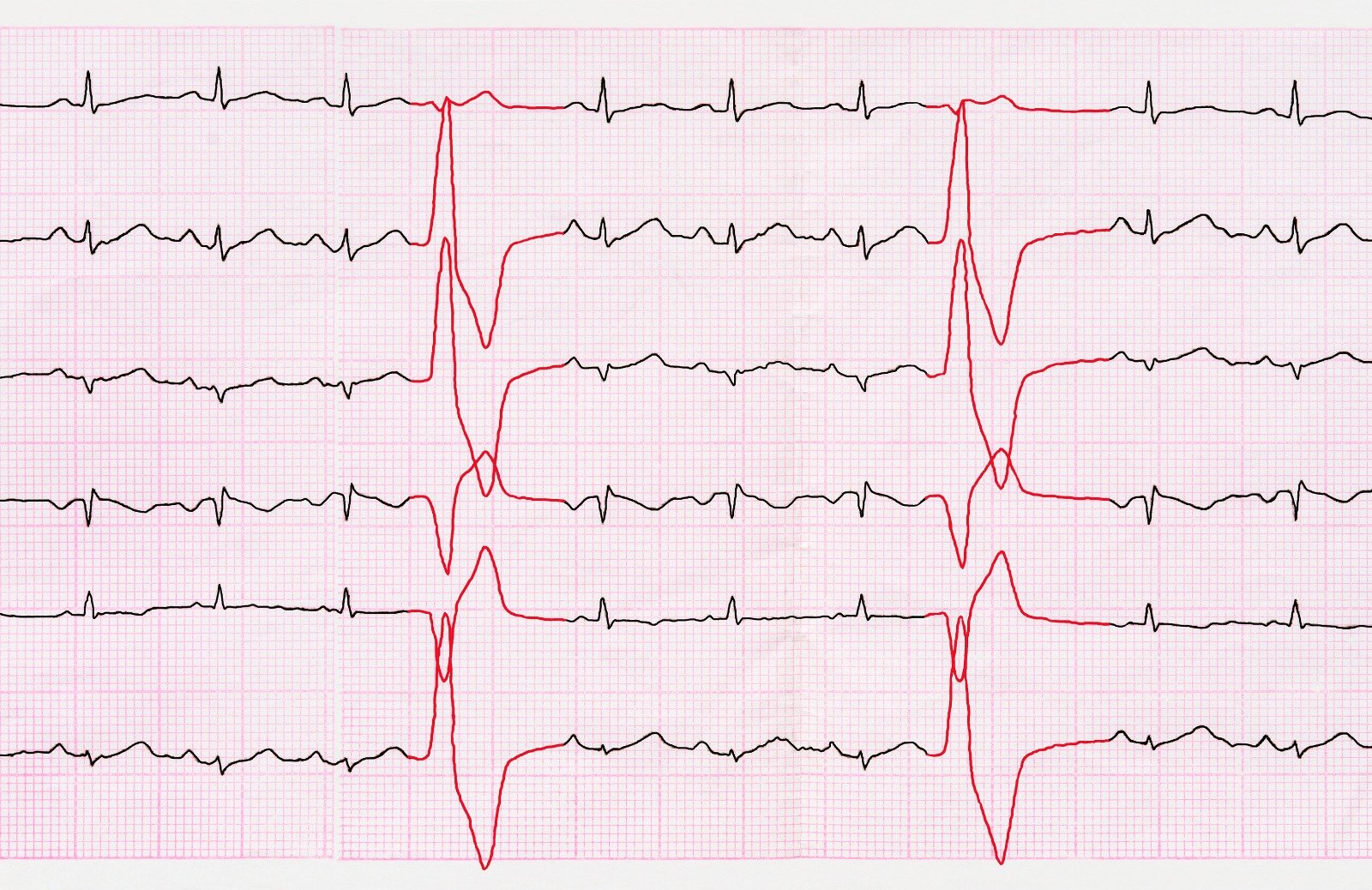
Electrocardiography (ECG)
- Should be performed on all patients, though not specific for HCM.
- Common abnormalities include repolarization changes, left-axis deviation, P-wave abnormalities, and inverted T waves.
Cardiac Catheterization
- Not mandatory for diagnosis but useful for assessing outflow obstruction, hemodynamics, and coronary anatomy.
Interventional options include transcatheter septal alcohol ablation.
Hemodynamic Abnormalities
- LV outflow gradient variability is characteristic of HCM.
- Factors influencing gradients include increased contractility, decreased preload, and decreased afterload.
Left Ventriculography
- Shows hypertrophied ventricle, outflow gradient, and mitral regurgitation.
- The LV cavity is often small with vigorous systolic ejection.
Electrophysiologic Studies (EPS)
- Used to identify conduction abnormalities and arrhythmias.
- Limited role in discovering mechanisms of sudden death.
Exercise Testing
- Important for risk stratification.
- Preferred over pharmacologic stress testing.
- Provides information on functional capacity, ischemia, arrhythmia, and obstruction.
Follow-up Considerations
Consider follow-up exercise testing to assess treatment efficacy.
Look for symptoms, blood pressure response, arrhythmias, and changes in LVOT gradient or mitral regurgitation during exercise.
Management & Treatment
Medical Therapy
- Medications include beta-blockers, calcium channel blockers, and, rarely, other agents like amiodarone.
- Mavacamten, an allosteric inhibitor of cardiac myosin, gained FDA approval for symptomatic obstructive HCM.
Invasive Procedures
- Cardiac catheterization for assessing outflow obstruction, hemodynamics, and coronary anatomy.
- Left ventricular myomectomy for severe symptoms and outflow gradient.
- Alcohol septal ablation as an alternative to myectomy.
Implantable Devices
- Implantable Cardioverter Defibrillator (ICD) for high-risk individuals.
- Pacemaker implantation may be considered for reducing LVOT gradient.
Heart Transplantation
Considered in advanced cases with refractory symptoms and NYHA class III or IV.
Atrial Fibrillation Management
- Use antiarrhythmic agents (e.g., amiodarone, sotalol) for AF control.
- Consider pulmonary vein catheter-based ablation for refractory cases.
Pregnancy Considerations
Generally safe with careful evaluation; cesarean delivery is usually not required.
Occupational Considerations
- Monitoring and restrictions on driving for certain individuals.
- Restrictions on commercial and private pilot licenses due to unpredictable risk.
Activity Recommendations
- Avoid strenuous exercise, especially in the presence of significant cardiac abnormalities.
- Consider cardiovascular screening before participation in competitive sports.
References:
- Facoi, S. N. S. D. M. F. F. (n.d.). Hypertrophic Cardiomyopathy: Background, Pathophysiology, Etiology. https://emedicine.medscape.com/article/152913
- Hypertrophic cardiomyopathy: Clinical manifestations, diagnosis, and evaluation. (n.d.). UpToDate. Retrieved December 20, 2023, from https://www.uptodate.com/contents/hypertrophic-cardiomyopathy-clinical-manifestations-diagnosis-and-evaluation


.webp)
.webp)
.webp)
.webp)
.webp)
.webp)